





Abstract
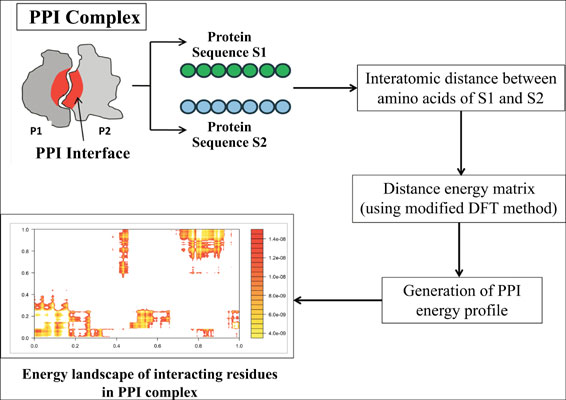
Background: Large-scale energy landscape characterization of protein-protein interactions (PPIs) is important to understand the interaction mechanism and protein-protein docking methods. The experimental methods for detecting energy landscapes are tedious and the existing computational methods require longer simulation time.
Objective: The objective of the present work is to ascertain the energy profiles at the interface regions in a rapid manner to analyze the energy landscape of protein-protein interactions.
Methods: The atomic coordinates obtained from the X-ray and NMR spectroscopy data are considered as inputs to compute cumulative energy profiles for experimentally validated protein-protein complexes. The energies computed by the program were comparable to the standard molecular dynamics simulations.
Results: The PPI Profiler not only enables rapid generation of energy profiles but also facilitates the detection of hot spot residue atoms involved therein.
Conclusion: The hotspot residues and their computed energies matched with the experimentally determined hot spot residues and their energies which correlated well by employing the MM/GBSA method. The proposed method can be employed to scan entire proteomes across species at an atomic level to study the key PPI interactions.
Keywords: Protein-protein interaction, energy profiles, DFT, energy landscape, hot spot residues, proteomes.
Graphical Abstract
[http://dx.doi.org/10.1109/TCBB.2016.2621042]
Related Journals

Anti-Cancer Agents in Medicinal Chemistry

Anti-Inflammatory & Anti-Allergy Agents in Medicinal Chemistry

Current Computer-Aided Drug Design

Current Bioactive Compounds

Current Cancer Drug Targets

Combinatorial Chemistry & High Throughput Screening

Current Cancer Therapy Reviews

Current Diabetes Reviews

Current Drug Safety

Current Drug Targets
Related Books

Drug Addiction Mechanisms in the Brain

Software and Programming Tools in Pharmaceutical Research

Objective Pharmaceutics: A Comprehensive Compilation of Questions and Answers for Pharmaceutics Exam Prep

Medicinal Chemistry of Drugs Affecting Cardiovascular and Endocrine Systems

Medicinal Plants, Phytomedicines and Traditional Herbal Remedies for Drug Discovery and Development against COVID-19

Advanced Pharmacy

Plant-derived Hepatoprotective Drugs

The Role of Chromenes in Drug Discovery and Development

New Avenues in Drug Discovery and Bioactive Natural Products

Practice and Re-Emergence of Herbal Medicine
 38
38 3
3 1
1 1
1