






Abstract
Background: Viruses have high mutation rates, facilitating rapid evolution and the emergence of new species, subspecies, strains and recombinant forms. Accurate classification of these forms is crucial for understanding viral evolution and developing therapeutic applications. Phylogenetic classification is typically performed by analyzing molecular differences at the genomic and sub-genomic levels. This involves aligning homologous proteins or genes. However, there is growing interest in developing alignment-free methods for whole-genome comparisons that are computationally efficient.
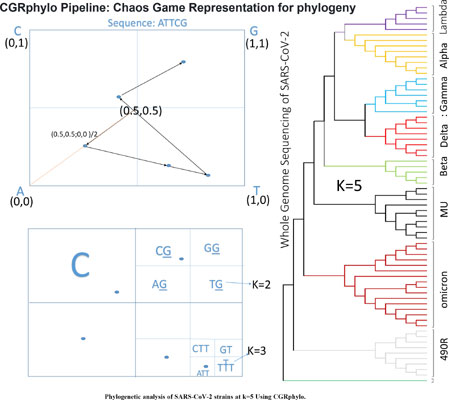
Methods: Here we elaborate on the Chaos Game Representation (CGR) method, based on concepts of statistical physics and free of sequence alignment assumptions. We adopt the CGR method for classification of the closely related clades/lineages A and B of the SARS-Corona virus 2019 (SARS-CoV-2), which is one of the fastest evolving viruses.
Results: Our study shows that the CGR approach can easily yield the SARS-CoV-2 phylogeny from the available whole genomes of lineage A and lineage B sequences. It also shows an accurate classification of eight different strains and the newly evolved XBB variant from its parental strains. Compared to alignment-based methods (Neighbour-Joining and Maximum Likelihood), the CGR method requires low computational resources, is fast and accurate for long sequences, and, being a K-mer based approach, allows simultaneous comparison of a large number of closely-related sequences of different sizes. Further, we developed an R pipeline CGRphylo, available on GitHub, which integrates the CGR module with various other R packages to create phylogenetic trees and visualize them.
Conclusion: Our findings demonstrate the efficacy of the CGR method for accurate classification and tracking of rapidly evolving viruses, offering valuable insights into the evolution and emergence of new SARS-CoV-2 strains and recombinants.
Keywords: CGR, alignment-free method, SARS-CoV-2, lineages A and B, XBB strain, phylogeny, CGRphylo, maximumlikelihood.
Graphical Abstract
[http://dx.doi.org/10.1371/journal.pbio.3000003] [PMID: 30102691]
[http://dx.doi.org/10.1016/j.anorl.2020.05.014] [PMID: 32773332]
[http://dx.doi.org/10.1038/s41597-020-0448-0] [PMID: 32210236]
[http://dx.doi.org/10.1016/S0140-6736(22)01924-9] [PMID: 36215997]
[http://dx.doi.org/10.1093/trstmh/traa025] [PMID: 32198918]
[http://dx.doi.org/10.1056/NEJMoa2001017] [PMID: 31978945]
[http://dx.doi.org/10.1016/S1473-3099(20)30120-1] [PMID: 32087114]
[http://dx.doi.org/10.1038/s41564-020-0709-x] [PMID: 32341570]
[http://dx.doi.org/10.1128/9781555819156.ch1]
[http://dx.doi.org/10.1128/br.35.3.235-241.1971] [PMID: 4329869]
[http://dx.doi.org/10.1038/nrg3186] [PMID: 22456349]
[http://dx.doi.org/10.1016/j.sbi.2006.04.004] [PMID: 16679011]
[http://dx.doi.org/10.1093/molbev/mst010] [PMID: 23329690]
[http://dx.doi.org/10.1002/0471250953.bi0313s48]
[http://dx.doi.org/10.1002/0471250953.bi0203s00]
[http://dx.doi.org/10.1038/s41467-018-04217-5] [PMID: 29765018]
[http://dx.doi.org/10.1073/pnas.0909377106] [PMID: 19805074]
[http://dx.doi.org/10.1073/pnas.0905115106] [PMID: 19553209]
[http://dx.doi.org/10.1186/s13059-017-1319-7] [PMID: 28974235]
[http://dx.doi.org/10.1186/1471-2105-11-322] [PMID: 20550657]
[http://dx.doi.org/10.1093/nar/gki541] [PMID: 15860779]
[http://dx.doi.org/10.1093/nar/18.8.2163] [PMID: 2336393]
[http://dx.doi.org/10.1093/nar/21.10.2487] [PMID: 8506142]
[http://dx.doi.org/10.1093/bioinformatics/17.5.429] [PMID: 11331237]
[http://dx.doi.org/10.4236/jbise.2009.28084]
[http://dx.doi.org/10.1186/1471-2105-11-S1-S26] [PMID: 20122198]
[http://dx.doi.org/10.1371/journal.pone.0206409] [PMID: 30427878]
[http://dx.doi.org/10.2807/1560-7917.ES.2017.22.13.30494] [PMID: 28382917]
[http://dx.doi.org/10.1093/molbev/mst012] [PMID: 23486614]
[http://dx.doi.org/10.1093/bioinformatics/bty633] [PMID: 30016406]
[http://dx.doi.org/10.1201/9781003279242]
Call for Papers in Thematic Issues
Advanced AI Techniques in Big Genomic Data Analysis
The thematic issue on "Advanced AI Techniques in Big Genomic Data Analysis" aims to explore the cutting-edge methodologies and applications of artificial intelligence (AI) in the realm of genomic research, where vast amounts of data pose both challenges and opportunities. This issue will cover a broad spectrum of AI-driven strategies, ...read more
Advanced Computational Algorithms and Artificial Intelligence in Clinical Pharmacogenomics
In the era of personalized medicine, understanding the relationship between genetics and drug response is crucial. This issue delves into innovative methodologies, leveraging deep computational analysis and artificial intelligence, to enhance the field of Clinical Pharmacogenomics. The interdisciplinary approach harnesses the power of advanced high-throughput genotyping technologies, sophisticated computational analysis, ...read more
Applications of Single-cell Sequencing Technology in Reproductive Medicine
Single cell sequencing (SCS) technology utilizes individual cells' genetic material to sequence their genome, transcriptome, and epigenetics at the molecular level. It offers insights into cell heterogeneity and enables the study of limited biological materials. Since its recognition as a valuable technique in 2011, single cell sequencing has yielded numerous ...read more
Big Data in Cancer Research
Cancer is a significant threat to human life and health, remaining a highly aggressive killer. It is a leading cause of death worldwide and represents a crucial medical issue for humanity. However, in the past decade, the effectiveness of new synthetic anticancer agents has not matched the current clinical speculation. ...read more
Related Journals










Related Books

Industrial Applications of Soil Microbes

Genome Editing in Bacteria (Part 2)

Aromatherapy: The Science of Essential Oils

In Vitro Propagation and Secondary Metabolite Production from Medicinal Plants: Current Trends (Part 2)

Micropropagation of Medicinal Plants

Software and Programming Tools in Pharmaceutical Research

Biotechnology and Drug Development for Targeting Human Diseases

In Vitro Propagation and Secondary Metabolite Production from Medicinal Plants: Current Trends (Part 1)

Molecular and Physiological Insights into Plant Stress Tolerance and Applications in Agriculture- Part 2

Micropropagation of Medicinal Plants
 18
18 2
2