





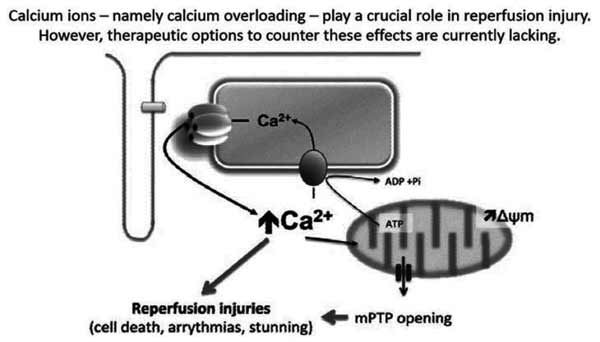
Abstract
Coronary Heart Disease (CHD) is the major mortality cause in the Western Hemisphere. Reinstituting blood flow in the acutely occluded coronary vessel became the standard intervention to prevent Myocardial Infarct (MI) progression. Ever since their conception, thrombolysis, Percutaneous Coronary Intervention (PCI) and Coronary Artery Bypass Grafting (CABG) have been at the forefront of CHD treatment, limiting MI size. However, it quickly became apparent that after a period of ischemia, reperfusion itself sets off a cascade of events leading to cell injury. It seems that cellular changes in the ischemic period, prime the cell for a loss of homeostasis once blood flow returns. Loss of calcium (Ca2+) regulation has been found to be a main culprit in both ischemia and reperfusion. Indeed, sarcoplasmic Ca2+ overload during reperfusion is related to hypercontracture, proteolysis and mitochondrial failure - the so-called Reperfusion Injury (RI). Ca2+ channels of the sarcolemma (SL) (L-Type Ca2+ Channels, Sodium / Calcium Exchanger) initiate Ca2+ flux and those of the Sarcoplasmic Reticulum (SR) (Ca2+ ATPase, Ca2+ release channel) sustain the rise in intracellular Ca2+ concentration. Ensuing interplay between Ca2+, SR, mitochondria, myofilaments and proteolytic cascades i.e. calpain activation, results in cell injury. Novel insight about this interplay and details about the extent by which each of these players contributes to the RI, may allow scientists to devise and design proper interventions that ultimately reduce RI in clinical practice. The present article reviews the literature about key subcellular players participating in the sustained rise of cardiac myocyte cytosolic Ca2+ during ischemia and reperfusion.
Keywords: Calcium, ischemia and reperfusion injury, L-type Ca2+ channel, Na+– Ca2+ exchanger, ryanodine receptor, sarco/endoplasmic reticulum Ca2+-ATPase, phospholamban, calpain.
Graphical Abstract
Related Journals

Anti-Cancer Agents in Medicinal Chemistry

Anti-Inflammatory & Anti-Allergy Agents in Medicinal Chemistry

Current Bioactive Compounds

Current Cancer Drug Targets

Combinatorial Chemistry & High Throughput Screening

Current Cancer Therapy Reviews

Current Diabetes Reviews

Current Drug Safety

Current Drug Targets

Current Drug Therapy
Related Books

Drug Addiction Mechanisms in the Brain

Botanicals and Natural Bioactives: Prevention and Treatment of Diseases

Software and Programming Tools in Pharmaceutical Research

Objective Pharmaceutics: A Comprehensive Compilation of Questions and Answers for Pharmaceutics Exam Prep

Medicinal Chemistry of Drugs Affecting Cardiovascular and Endocrine Systems

Frontiers In Medicinal Chemistry

Advanced Pharmacy

Plant-derived Hepatoprotective Drugs

Frontiers in Natural Product Chemistry

The Role of Chromenes in Drug Discovery and Development
 85
85 7
7 2
2 1
1