





Abstract
Background: Atrial fibrillation (AF) is one of the most common heart arrhythmic disorders all over the world. However, it is worth noting that the mechanism underlying AF is still dimness.
Methods: In this study, we implemented a series of bioinformatics methods to explore the mechanisms of lncRNAs underlying AF pathogenesis. The present study analyzed the public datasets (GSE2240 and GSE115574) to identify differentially expressed long non-coding RNAs (lncRNAs) and mRNAs in the progression of AF.
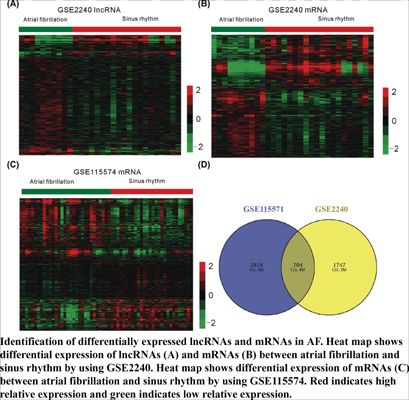
Results: Totally, 71 differentially expressed lncRNAs and 390 DEGs were identified in AF.Next, we performed bioinformatics analyses to explore the functions of lncRNAs in AF. Gene Ontology (GO) analysis indicated that differentially expressed lncRNAs were involved in regulating multiple key biological processes, such as cell cycle and signal transduction. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis demonstrated these lncRNAs were associated with the regulation of MAPK and Wnt signaling pathways. Eight lncRNAs (RP5-1154L15.2, RP11- 339B21.15, RP11-448A19.1, RP11-676J12.4, LOC101930415, MALAT1, NEAT1, and PWAR6) were identified to be key lncRNAs and widely co-expressed with a series of differentially expressed genes (DEGs).
Conclusion: Although further validation was still needed, our study may be helpful to elucidate the mechanisms of lncRNAs underlying AF pathogenesis and providing further insight into identifying novel biomarkers for AF.
Keywords: Long non-coding RNA, atrial fibrillation, co-expression network analysis, expression profiling, gene, clustering.
Graphical Abstract










Related Books

Industrial Applications of Soil Microbes

Genome Editing in Bacteria (Part 2)

Aromatherapy: The Science of Essential Oils

In Vitro Propagation and Secondary Metabolite Production from Medicinal Plants: Current Trends (Part 2)

Micropropagation of Medicinal Plants

Software and Programming Tools in Pharmaceutical Research

Biotechnology and Drug Development for Targeting Human Diseases

In Vitro Propagation and Secondary Metabolite Production from Medicinal Plants: Current Trends (Part 1)

Molecular and Physiological Insights into Plant Stress Tolerance and Applications in Agriculture- Part 2

Micropropagation of Medicinal Plants
 40
40