






Abstract
Background: De novo peptide sequencing is one of the key technologies in proteomics, which can extract peptide sequences directly from tandem mass spectrometry (MS/MS) spectra without any protein databases. Since the accuracy and efficiency of de novo peptide sequencing can be affected by the quality of the MS/MS data, the DeepNovo method using deep learning for de novo peptide sequencing is introduced, which outperforms the other state-of-the-art de novo sequencing methods.
Objective: For superior performance and better generalization ability, additional ion types of spectra should be considered and the model of DeepNovo should be adaptive.
Methods: Two improvements are introduced in the DeepNovo A+ method: a_ions are added in the spectral analysis, and the validation set is used to automatically determine the number of training epochs.
Results: Experiments show that compared to the DeepNovo method, the DeepNovo A+ method can consistently improve the accuracy of de novo sequencing under different conditions.
Conclusion: By adding a_ions and using the validation set, the performance of de novo sequencing can be improved effectively.
Keywords: MS/MS spectra, de novo peptide sequencing, DeepNovo, deep learning, validation set, fragment ions.
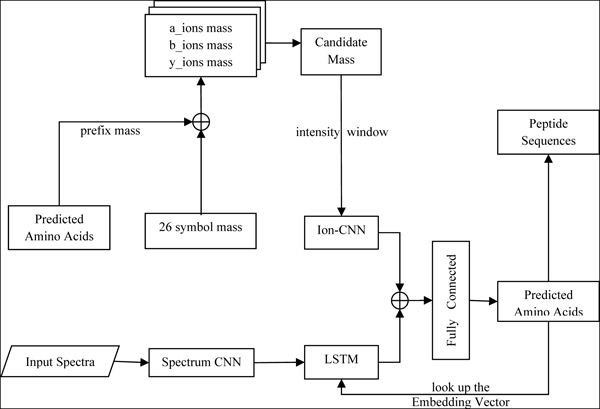
Graphical Abstract
[http://dx.doi.org/10.1021/bi00379a001] [PMID: 3567166]
[http://dx.doi.org/10.1073/pnas.0800585105] [PMID: 18635686]
[http://dx.doi.org/10.1002/pmic.201400349] [PMID: 25487722]
[PMID: 20013367]
[http://dx.doi.org/10.1016/1044-0305(94)80016-2] [PMID: 24226387]
[http://dx.doi.org/10.1002/(SICI)1522-2683(19991201)20:18<3551:AID-ELPS3551>3.0.CO;2-2] [PMID: 10612281]
[http://dx.doi.org/10.1002/rcm.1198] [PMID: 14558131]
[http://dx.doi.org/10.1093/bioinformatics/bth092] [PMID: 14976030]
[http://dx.doi.org/10.1021/pr101065j] [PMID: 21254760]
[http://dx.doi.org/10.1093/bioinformatics/bth186] [PMID: 15044235]
[http://dx.doi.org/10.1002/rcm.3173] [PMID: 17702057]
[http://dx.doi.org/10.1038/ncomms6277] [PMID: 25358478]
[http://dx.doi.org/10.1002/0471250953.bi1320s40]
[http://dx.doi.org/10.1586/epr.11.54] [PMID: 21999834]
[http://dx.doi.org/10.1002/(SICI)1097-0231(19970615)11:9<1067:AID-RCM953>3.0.CO;2-L] [PMID: 9204580]
[http://dx.doi.org/10.1089/106652799318300] [PMID: 10582570]
[http://dx.doi.org/10.1002/rcm.1196] [PMID: 14558135]
[http://dx.doi.org/10.1021/ac0508853] [PMID: 16285674]
[http://dx.doi.org/10.1021/ac048788h] [PMID: 15858974]
[http://dx.doi.org/10.1021/pr060271u] [PMID: 17203955]
[http://dx.doi.org/10.1021/pr100182k] [PMID: 20329752]
[http://dx.doi.org/10.1021/pr3006843] [PMID: 23272783]
[http://dx.doi.org/10.1093/bioinformatics/btt338] [PMID: 23766417]
[http://dx.doi.org/10.1007/s13361-015-1204-0] [PMID: 26122521]
[http://dx.doi.org/10.1109/MSP.2012.2205597]
[http://dx.doi.org/10.1073/pnas.1705691114] [PMID: 28720701]
[http://dx.doi.org/10.1021/acs.jproteome.6b00647] [PMID: 27966978]
[http://dx.doi.org/10.1038/s41587-019-0067-5] [PMID: 30936560]










Related Books

In Vitro Propagation and Secondary Metabolite Production from Medicinal Plants: Current Trends (Part 2)

Micropropagation of Medicinal Plants

Software and Programming Tools in Pharmaceutical Research

Biotechnology and Drug Development for Targeting Human Diseases

In Vitro Propagation and Secondary Metabolite Production from Medicinal Plants: Current Trends (Part 1)

Molecular and Physiological Insights into Plant Stress Tolerance and Applications in Agriculture- Part 2

Micropropagation of Medicinal Plants

Genome Editing in Bacteria (Part 1)

Data Science for Agricultural Innovation and Productivity

Animal Models In Experimental Medicine
 24
24