






Abstract
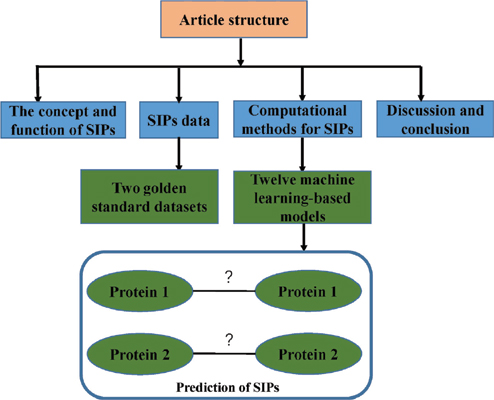
Self-Interacting Proteins (SIPs), whose two or more copies can interact with each other, have significant roles in cellular functions and evolution of Protein Interaction Networks (PINs). Knowing whether a protein can act on itself is important to understand its functions. Previous studies on SIPs have focused on their structures and functions, while their whole properties are less emphasized. Not surprisingly, identifying SIPs is one of the most important works in biomedical research, which will help to understanding the function and mechanism of proteins. It is worth noting that high throughput methods can be used for SIPs prediction, but can be costly, time consuming and challenging. Therefore, it is urgent to design computational models for the identification of SIPs. In this review, the concept and function of SIPs were introduced in detail. We further introduced SIPs data and some excellent computational models that have been designed for SIPs prediction. Specially, the most existing approaches were developed based on machine learning through carrying out different extract feature methods. Finally, we discussed several difficult problems in developing computational models for SIPs prediction.
Keywords: Self-interacting proteins, computational models, protein interaction, machine learning, biomedical research, cellular functions.
Graphical Abstract
[http://dx.doi.org/10.3389/fgene.2019.00090] [PMID: 30881376]
[http://dx.doi.org/10.1021/pr050331g] [PMID: 16457597]
[http://dx.doi.org/10.1016/j.tibs.2004.09.006] [PMID: 15501681]
[http://dx.doi.org/10.18632/oncotarget.12517] [PMID: 27732957]
[http://dx.doi.org/10.1186/s13321-017-0233-z] [PMID: 29086182]
[http://dx.doi.org/10.1074/jbc.M414440200] [PMID: 15691829]
[http://dx.doi.org/10.1038/sj.onc.1206204] [PMID: 12618752]
[http://dx.doi.org/10.1038/328834a0] [PMID: 3627230]
[http://dx.doi.org/10.1101/gr.128819.111] [PMID: 22194470]
[http://dx.doi.org/10.1016/j.celrep.2013.08.028] [PMID: 24075989]
[http://dx.doi.org/10.1093/bib/bbx142] [PMID: 29165544]
[http://dx.doi.org/10.1093/nar/gki678] [PMID: 15983135]
[http://dx.doi.org/10.1093/bioinformatics/btq510] [PMID: 20817744]
[PMID: 25348405]
[http://dx.doi.org/10.1093/nar/gkq1116]
[http://dx.doi.org/10.1093/nar/28.1.289] [PMID: 10592249]
[http://dx.doi.org/10.1093/nar/gkt1115] [PMID: 24234451]
[http://dx.doi.org/10.1093/nar/gku1091] [PMID: 25378329]
[http://dx.doi.org/10.1093/nar/gks1147] [PMID: 23180781]
[http://dx.doi.org/10.1016/j.jtbi.2017.08.009] [PMID: 28802824]
[http://dx.doi.org/10.7150/ijbs.23817] [PMID: 29989064]
[http://dx.doi.org/10.1186/s12918-018-0647-x] [PMID: 30577794]
[http://dx.doi.org/10.3390/ijms20040930] [PMID: 30795499]
[http://dx.doi.org/10.1074/mcp.M112.021790] [PMID: 23422585]
[http://dx.doi.org/10.1109/TCBB.2017.2649529] [PMID: 28092572]
[http://dx.doi.org/10.1039/C6MB00599C] [PMID: 27759121]
[http://dx.doi.org/10.1007/s00726-016-2226-z] [PMID: 27074717]
[http://dx.doi.org/10.1016/j.compbiolchem.2017.03.009] [PMID: 28396055]
[http://dx.doi.org/10.1109/TNB.2015.2429672] [PMID: 26011889]
[http://dx.doi.org/10.1371/journal.pcbi.1006418] [PMID: 30142158]
[http://dx.doi.org/10.1093/bib/bbx130] [PMID: 29045685]
[http://dx.doi.org/10.1093/bioinformatics/bty333] [PMID: 29701758]
[PMID: 27345524]
[http://dx.doi.org/10.1093/bib/bbv066] [PMID: 26283676]










Related Books

Industrial Applications of Soil Microbes

Genome Editing in Bacteria (Part 2)

Aromatherapy: The Science of Essential Oils

In Vitro Propagation and Secondary Metabolite Production from Medicinal Plants: Current Trends (Part 2)

Micropropagation of Medicinal Plants

Software and Programming Tools in Pharmaceutical Research

Biotechnology and Drug Development for Targeting Human Diseases

In Vitro Propagation and Secondary Metabolite Production from Medicinal Plants: Current Trends (Part 1)

Molecular and Physiological Insights into Plant Stress Tolerance and Applications in Agriculture- Part 2

Micropropagation of Medicinal Plants
 23
23 1
1